My first simulation#
As introduction to the gamdpy package let us setting up a simulation
of a Lennard-Jones FCC crystal in the constant \(NVT\) ensemble.
For a short summary, see the minimal.py example script. Also, the line sim = gp.get_default_simulation() will create a simulation object similar to the below.
If gamdpy is installed correctly, the following package should be imported without any errors. It is recommended to import the package as gp.
import gamdpy as gp
*We will also import NumPy for numerical calculations and Matplotlib for plotting.
import numpy as np # For numerical operations
import matplotlib.pyplot as plt # For plotting
Initial particle positions#
We wish to set up a configuration of a FCC lattice with \(8 \times 8 \times 8\) unit cells, with a density of \(\rho = 0.973\). First we create an empty configuration object.
configuration = gp.Configuration(D=3)
Information for a few crystal unit cells are avalible with the gamdpy package. For example, the FCC unit cell is stored as rp.unit_cells.FCC.
gp.unit_cells.FCC
{'fractional_coordinates': [[0.0, 0.0, 0.0],
[0.5, 0.5, 0.0],
[0.5, 0.0, 0.5],
[0.0, 0.5, 0.5]],
'lattice_constants': [1.0, 1.0, 1.0]}
We use the make_lattice method to assign positions and a simulation box to the configuration object.
# Setup configuration: FCC Lattice
configuration.make_lattice(
unit_cell=gp.unit_cells.FCC,
cells=[8, 8, 8],
rho=0.973
)
The positions can be accessed with ['r']. All vector properties, like the positions, are NumPy arrays.
positions = configuration['r']
type(positions)
numpy.ndarray
To confirm that the positions are as expected, we can visualize the configuration using Matplotlib.
# Make 3D plot using matplotlib
fig = plt.figure()
ax = fig.add_subplot(111, projection='3d')
ax.set_title('Initial configuration')
ax.scatter(positions[:,0], positions[:,1], positions[:,2])
ax.set_xlabel('x')
ax.set_ylabel('y')
ax.set_zlabel('z')
plt.show()
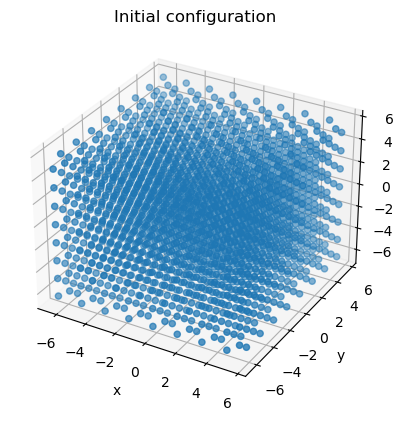
The configuration object also contains a simulation box. Here we use an orthorhombic box. The box lengths can be accessed as seen below.
configuration.simbox.get_lengths() # Box lengths
array([12.815602, 12.815602, 12.815602], dtype=float32)
Masses and initial velocities#
We set all particle masses to one (the value is broadcast to all particles).
configuration['m'] = 1.0 # Set all masses to 1.0
Note that configuration['m'] refer to an NumPy array. For convenience, we above used a float to set all masses to the same value. This value will automatically be broadcast to all particles.
type(configuration['m']) # configuration['m'] is a NumPy array
numpy.ndarray
After masses are set, we can assign random velocities corresponding to an initial kinetic temperature \(T = 0.7\).
temperature = 0.7
configuration.randomize_velocities(temperature=temperature)
To confirm that the initial velocity distribution is as expected, we can compare it the the theoretical Maxwell-Boltzmann distribution.
plt.figure()
# Histogram of particle speeds
vel = configuration['v']
speeds = np.linalg.norm(vel, axis=1)
bins = np.linspace(0, 5, 25)
plt.hist(speeds, bins=bins, density=True, label='Simulation')
# Theoretical Maxwell-Boltzmann distribution
v = np.linspace(0, 5, 100)
maxwell_boltzmann = (2*np.pi*temperature)**(-3/2)*4*np.pi*v**2*np.exp(-v**2/(2*temperature))
plt.plot(v, maxwell_boltzmann, label='Maxwell-Boltzmann', color='r')
# Plot settings
plt.xlabel(r'Particle speeds, $|v|$')
plt.ylabel(r'Probability density, $P(|v|)$')
plt.xlim(0, None)
plt.legend()
plt.show()
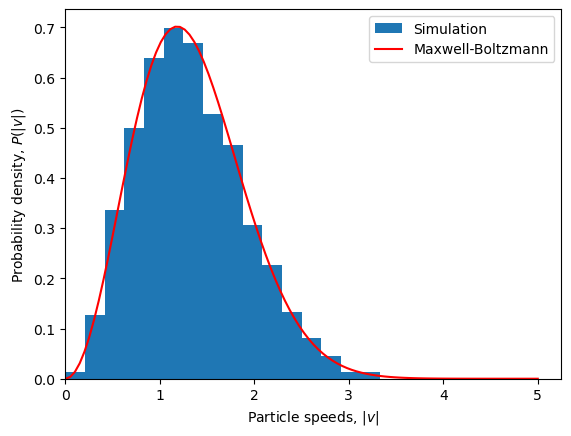
Pair potential#
Next, we set up the Lennard-Jones pair potential. We use the 12-6 Lennard-Jones potential
(the substript refer to a “truncation” at \(\infty\)). Here \(\sigma\) (sigma) is length, and \(\varepsilon\) (epsilon) is an energy. We will use Lennard-Jones units, where \(\sigma = 1\) and \(\varepsilon = 1\).
Several pair potentials functions are implemented in gamdpy, and the above 12-6 Lennard-Jones potential is implemented as gp.LJ_12_6_sigma_epsilon. This is a Python function that takes distances, parameters as input and returns the potential energy, the force and the second derivative of the potential energy.
Let us confirm values are as expected in the minimum of the potential:
pair_func = gp.LJ_12_6_sigma_epsilon # Pair potential function
r_min = 2**(1/6)
u_min, f_min, curvature_min = pair_func(r_min, (1.0, 1.0, np.inf))
u_min, f_min, curvature_min
(-1.0, -2.1148430366511475e-15, 57.146437870855145)
To allow for a \(O(N)\) algorithm for force calculations, we truncate the pair potential at radius of \(r_c = 2.5\sigma\) (cutoff). The potential is shifted to zero at the cutoff radius, so the actual potential is
For this, we can use gp.apply_shifted_potential_cutoff to modify the pair potential.
pair_func = gp.apply_shifted_potential_cutoff(gp.LJ_12_6_sigma_epsilon)
Let us plot the pair potential, so confirm that it looks as we expect.
sigma, epsilon, cutoff = 1.0, 1.0, 2.5
pair_distances = np.linspace(0.95, 3.0, 128)
pair_energies = [pair_func(dist, [sigma, epsilon, cutoff])[0] for dist in pair_distances]
plt.figure()
plt.plot(pair_distances, pair_energies)
plt.xlabel(r'Pair distance, r')
plt.ylabel(r'Pair energy, $v(r)$')
plt.show()
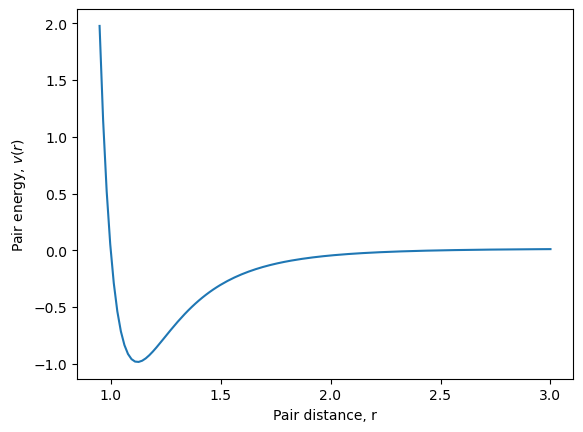
We can now create an interaction objects using this pair potential function we made.
pair_pot = gp.PairPotential(pair_func, params=[sigma, epsilon, cutoff], max_num_nbs=1000)
Finally, we create a list will all the interactions in our simulations. In this simulation, we only have one, but later we may want to include more like bonds, walls, or gravity.
interactions = [pair_pot, ]
Integrator#
Next, create an integrator object. These are available in the submodule gamdpy.integrators.
Here, we chose to use and implementation of the Nosé-Hoover thermostat discretion using the Leap-Frog scheme (others are available).
We set a time-step of \(dt=0.005\) and a thermostat relaxation time to \(\tau = 0.2\) (both in Lennard-Jones units).
# Setup integrator: Nosé-Hoover NVT, Leap-frog algorithm
integrator = gp.integrators.NVT(temperature=temperature, tau=0.2, dt=0.005)
Runtime actions#
We typically want to perform some actions during the simulations. These ‘runtime actions’ are collected in a list.
The
gp.TrajectorySaversaves positions using a logarithmic timescale in each timeblock. This can later be used to calculate properties such as the mean-square displacement etc.The
gp.ScalarSaversave scalar properties like energy everysteps_between_output=16timestep.The
gp.RestartSaversaves a ‘restart’, at the beginning of each timeblock. A restart contains all the information about a configuration, so that a new simulation can be initialized from this.During long-time simulations, there might be a drift of the total momentum due to floating point rounding erros. Thus, the
gp.MomentumResetreset the total momentum everysteps_between_reset=100timestep. All the runtime actions are collected in a list.
steps_between_output = 16
runtime_actions = [
gp.TrajectorySaver(),
gp.ScalarSaver(steps_between_output=steps_between_output),
gp.RestartSaver(),
gp.MomentumReset(steps_between_reset=100),
]
The Simulation object#
We now have all the components to set up a simulation object.
An configuration object (particle positions, velocities, masses, …).
A list of interactions (here the Lennard-Jones potential).
An integrator object (here NVT using the Nosé-Hoover thermostat).
A list of runtime actions
For the simulation object we also specify that we want num_timeblocks=32 time blocks of steps_per_timeblock=1024 steps. We also specify that we want to store the simulation data in a file with storage='LJ_T0.70.h5'. Change to storage='memory' if you do not want to save a file to the disk.
# Setup Simulation
sim = gp.Simulation(configuration, interactions, integrator, runtime_actions,
num_timeblocks=32,
steps_per_timeblock=1024,
storage='LJ_T0.70.h5')
Running the simulation#
Finally, we run the simulation using the sim.run_timeblocs iterator that will loop over all the timeblocs.
Below we print the status of the simulation after each timeblock.
for timeblock in sim.run_timeblocks():
print(sim.status(per_particle=True))
print(sim.summary())
timeblock= 0 time= 5.120 U= -6.288 W= 0.698 K= 1.058
timeblock= 1 time= 10.240 U= -6.278 W= 0.755 K= 1.068
timeblock= 2 time= 15.360 U= -6.270 W= 0.806 K= 1.026
timeblock= 3 time= 20.480 U= -6.277 W= 0.767 K= 1.036
timeblock= 4 time= 25.600 U= -6.292 W= 0.677 K= 1.053
timeblock= 5 time= 30.720 U= -6.268 W= 0.805 K= 1.075
timeblock= 6 time= 35.840 U= -6.326 W= 0.497 K= 1.033
timeblock= 7 time= 40.960 U= -6.286 W= 0.723 K= 1.075
timeblock= 8 time= 46.080 U= -6.295 W= 0.668 K= 1.035
timeblock= 9 time= 51.200 U= -6.305 W= 0.617 K= 1.077
timeblock= 10 time= 56.320 U= -6.268 W= 0.801 K= 1.056
timeblock= 11 time= 61.440 U= -6.278 W= 0.758 K= 1.039
timeblock= 12 time= 66.560 U= -6.279 W= 0.748 K= 1.021
timeblock= 13 time= 71.680 U= -6.313 W= 0.562 K= 1.022
timeblock= 14 time= 76.800 U= -6.269 W= 0.820 K= 1.046
timeblock= 15 time= 81.920 U= -6.274 W= 0.791 K= 1.023
timeblock= 16 time= 87.040 U= -6.287 W= 0.713 K= 1.042
timeblock= 17 time= 92.160 U= -6.271 W= 0.806 K= 1.055
timeblock= 18 time= 97.280 U= -6.288 W= 0.709 K= 1.069
timeblock= 19 time= 102.400 U= -6.274 W= 0.779 K= 1.042
timeblock= 20 time= 107.520 U= -6.270 W= 0.790 K= 1.036
timeblock= 21 time= 112.640 U= -6.293 W= 0.680 K= 1.046
timeblock= 22 time= 117.760 U= -6.278 W= 0.763 K= 1.063
timeblock= 23 time= 122.880 U= -6.290 W= 0.699 K= 1.054
timeblock= 24 time= 128.000 U= -6.275 W= 0.787 K= 1.082
timeblock= 25 time= 133.120 U= -6.282 W= 0.733 K= 1.063
timeblock= 26 time= 138.240 U= -6.310 W= 0.571 K= 1.050
timeblock= 27 time= 143.360 U= -6.281 W= 0.742 K= 1.069
timeblock= 28 time= 148.480 U= -6.275 W= 0.769 K= 1.030
timeblock= 29 time= 153.600 U= -6.271 W= 0.803 K= 1.041
timeblock= 30 time= 158.720 U= -6.255 W= 0.875 K= 1.062
timeblock= 31 time= 163.840 U= -6.284 W= 0.722 K= 1.027
Particles : 2048
Steps : 32 * 1024 = 32_768
Total run time (incl. time spent between timeblocks): 4.19 s ( TPS: 7.83e+03 )
Simulation time (excl. time spent between timeblocks): 1.28 s ( TPS: 2.56e+04 )
The sim.status() and sim.summary() methods prints some basic information.
Analysis of simulation#
The recomended workflow is to use seperate notebooks and/or python scripts to analyse the results from a simulation, utlilizing the data stored in a h5 file (in this case: LJ_T0.70.h5).
Examples of doing such data analysis are: post_analysis.ipynb, analyze_dynamics.py, analyze_structure.py, analyze_thermodynamics.py
In the following, we perform some data analysis, utlizing that the simulation data stored in the h5 file also is accecible immediatly in sim.output.
First, we extract some thermodynamic quantities stored in the simulation object.
# Extract potential energy, virial and kinetic energy
U, W, K = gp.ScalarSaver.extract(sim.output, ['U', 'W', 'K'], per_particle=False)
type(U)
numpy.ndarray
The variables U, W and K are NumPy arrays with the potential energy, virial and kinetic energy, respectively, for selected timesteps. The number of stored timestep can be found with len(U).
len(U)
2048
For plotting below, we get the times related to when the scalar quantities were stored (in Lennard-Jones units).
times = gp.ScalarSaver.get_times(sim.output)
len(times)
2048
We can use Matplotlib to plot the potential energy per particle as a function of time. We use sim.configuration.N to get the number of particles.
N = sim.configuration.N # Number of particles
plt.figure()
plt.plot(times, U/N)
plt.xlabel(r'Time, $t$ [$\sigma\sqrt{m/\varepsilon}$]') # Time in LJ units
plt.ylabel(r'Potential energy per particle, $U$ [$\varepsilon$]') # Energy in LJ units
plt.xlim(0, None)
plt.show()
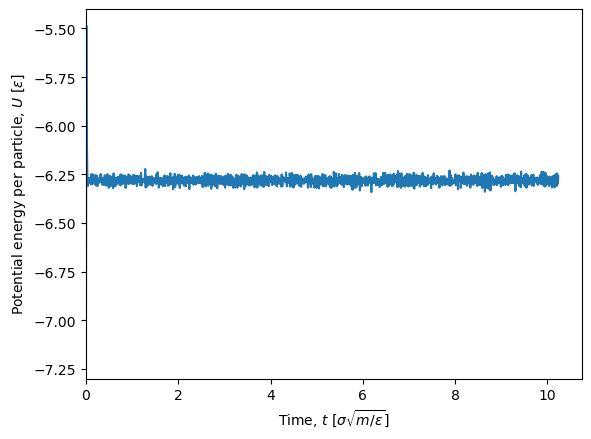
The first part of the simulation show large fluctuations in the potential energy since the system is not yet equilibrated (it was setup in a perfect crystal). Since we are not interested in the equilibration phase, we can discard the fir st timeblock by using the first_block=1 argument in the extract_scalars function. We then need to recalculate the time array.
U, W, K = gp.ScalarSaver.extract(sim.output, ['U', 'W', 'K'], per_particle=False, first_block=1)
times = gp.ScalarSaver.get_times(sim.output, first_block=1)
plt.figure()
plt.plot(times, U/N, label='Potential energy per particle')
plt.xlabel(r'Time, $t$ [$\sigma\sqrt{m/\varepsilon}$]') # Time in LJ units
plt.ylabel(r'Energy [$\varepsilon$]') # Energy in LJ units
plt.legend()
plt.show()
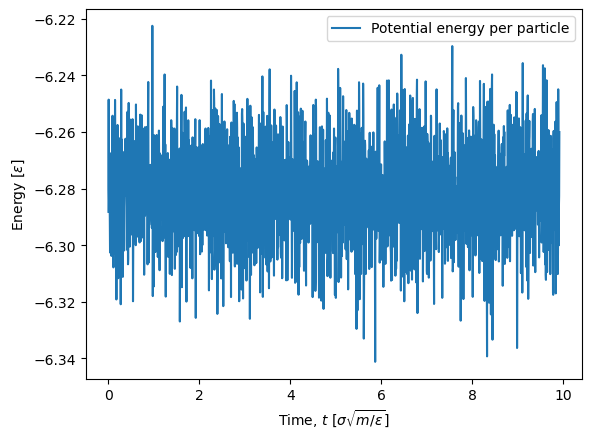
After we have ensured that we have an equilibrium simulation, the expectation value of the potential energy per particle can be found with the np.mean NumPy function.
np.mean(U/N)
-6.281355
In an \(NVT\) simulation the pressure is often an important quantity, but is not stored in the simulation data directly. The pressure can be calculated from the virial, \(W\). Recall,
where \(p\) is the pressure, \(V\) is the volume, \(N\) is the number of particles, \(k\) is the Boltzmann constant and \(T\) is the temperature. The volume can be calculated from the box lengths. In Lennard-Jones units boltzmann constant is \(k_B = 1\). Below we use this to calculate the pressure.
V = sim.configuration.get_volume() # Volume
dof = 3*N - 3 # Degrees of freedom
T_kin = 2*K/dof # Kinetic temperature
p = (N*T_kin + W)/V # Pressure
np.mean(p) # Mean pressure
1.4021976
Concluding remarks#
Congratulations! You have now set up and executed your first simulation with gamdpy.
Simulation data have been stored to the disk for later analysis.